The purpose of pedFamilias is to facilitate data
exchanging between the pedsuite packages and the Familias software for forensic kinship
analysis. The main functions are readFam() and
writeFam() for reading and writing .fam files,
which are used by Familias.
The readFam() function also supports files written with
the DVI module of Familias, and also pure database files
without pedigree information.
These functions were originally part of forrel, but were split out to streamline maintenance and provide a more lightweight import for other packages otherwise independent of forrel.
Install pedFamilias from CRAN as follows:
install.packages("pedFamilias")Alternatively, install the development version from GitHub:
# install.packages("remotes")
remotes::install_github("magnusdv/pedFamilias")library(pedFamilias)For a simple illustration of pedFamilias we read the
example file paternity.fam shipped with the package:
fam = system.file("extdata", "paternity.fam", package = "pedFamilias")
peds = readFam(fam)
#> Familias version: 3.3.1
#> Read DVI: No
#>
#> Number of individuals (excluding 'extras'): 3
#> Individual 'AF': Genotypes for 10 markers read
#> Individual 'MO': Genotypes for 0 markers read
#> Individual 'CH': Genotypes for 10 markers read
#>
#> Number of pedigrees: 2
#> Pedigree 'H1' (0 extra females, 0 extra males)
#> Pedigree 'H2' (0 extra females, 0 extra males)
#>
#> Database: unknown
#> Number of loci: 10
#> D3S1358: 12 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> TH01: 10 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> D21S11: 26 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> D18S51: 23 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> PENTA_E: 21 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> D5S818: 9 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> D13S317: 9 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> D7S820: 19 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> D16S539: 9 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#> CSF1PO: 11 alleles, mut model (M/F) = step-ext, rate = 0.002/0.001, range = 0.1, rate2 = 1e-06
#>
#> Converting to `ped` formatHere are the pedigrees, including genotypes for the first marker:
plotPedList(peds, hatched = typedMembers, marker = 1)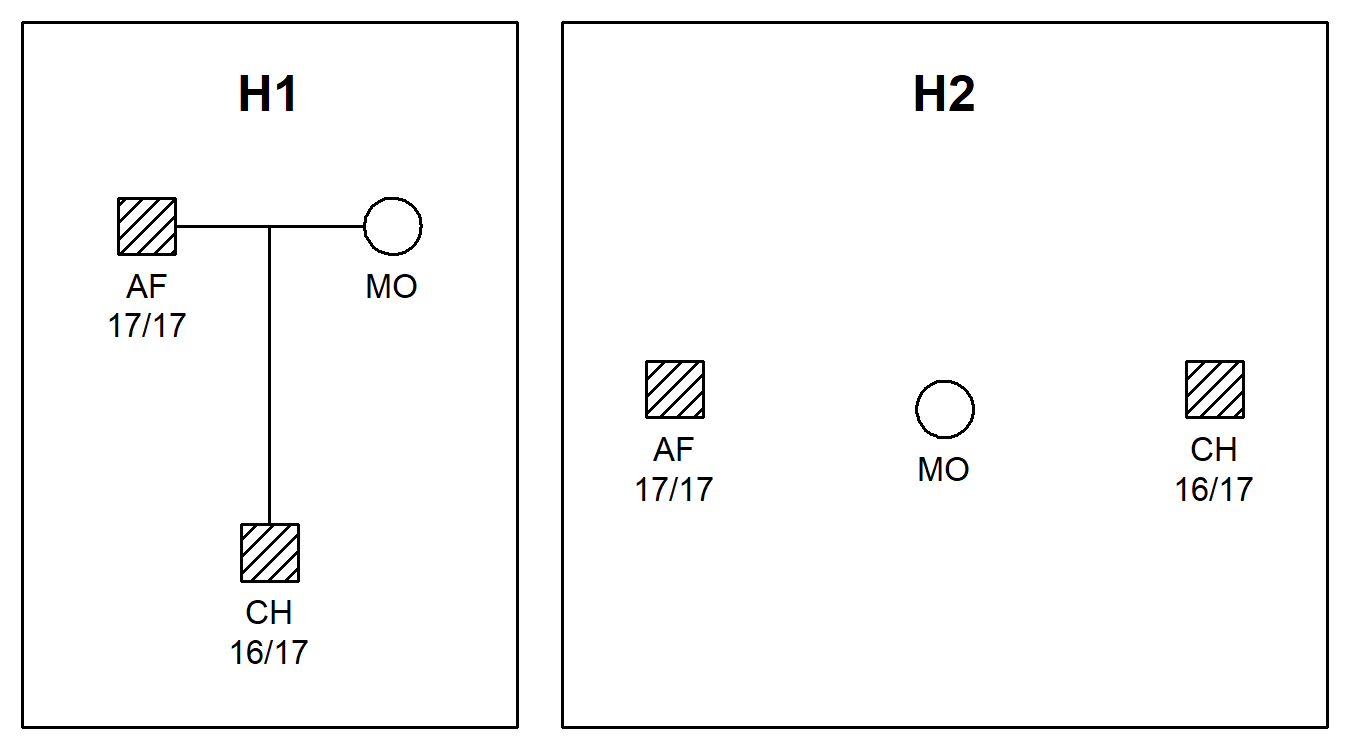
Further analysis of the data may be carried out with the forrel package. For instance, the following command computes the likelihood ratio comparing the two hypotheses:
forrel::kinshipLR(peds)
#> H1:H2 H2:H2
#> 760.3445 1.0000